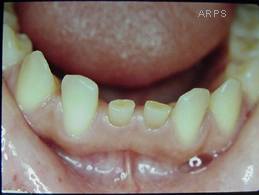
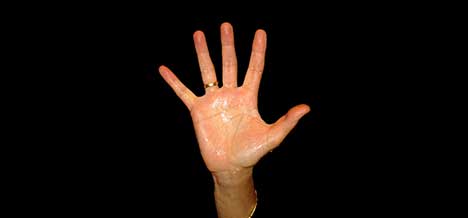
Esta enfermedad se caracteriza porque el enfero posee unas escamas grandes y gruesas que aparecen en toda la piel y tienen los párpados volteados (Por lo que estos se ven completamente rojos). Esta enfermedad está asociada generalmente a deformaciones faciales muy características, y, en ocasiones, a otras deformaciones en el tórax.
Esta enfermedad es provocada por una alteración en la queratinización, en la que su mecanismo fisiopatológico se desconoce, aunque se piensa que puede ser provocada por anomalias de los lípidos cutáneos, lo que tiene como consecuencia una hiperqueratosis folicular masiva.
Las principales características son que aparecen unas fisuras profundas e irregulares que cubren todo el cuerpo, eversión de los párpados, inversión hacia fuera de los labios, desarrollo incompleto o defectuoso de orejas nariz y dedos y los recién nacidos adoptan una postura semiflexionada.
Esta enfermedad provoca, entre otras cosas, sepsis, que es una infección o contaminación generalizada, y deshidratación, que conducen a un aumento anormal de sodio en la sangre y a una malnutrición. Esta enfermedad también provoca dificultados respiratorias y alimenticias.
La mayoría de los pacientes de la Ictiosis Arlequín fallecen a los pocos días del nacimiento, aunque existen casos excepcionales en los que la persona llega a la adolescencia. Estos pacientes necesitan tratamientos especiales que están aún en investigación.